
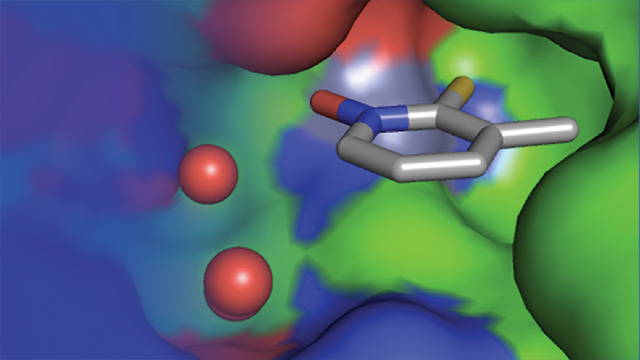
 This fragment binds a protein’s active site. Image courtesy of Seth Cohen
This fragment binds a protein’s active site. Image courtesy of Seth Cohen
In the pharmaceutical industry, the “batting average” for taking a compound from discovery to a marketed therapy leaves a lot to be desired. Consequently, pharmaceutical scientists keep looking for new techniques. While these scientists are thinking big, something smaller may be needed—particularly when it comes to searching for new lead compounds to test as medicines. A promising lead candidate should be specific and selective, targeting one site and only that site. That’s not so easy to find, because many molecules cling to too many others, and bigger molecules have more potential binding regions. Instead of working with those big molecules, pharmaceutical scientists can work with fragments— smaller pieces of potential drugs. Fragment-based drug discovery (FBDD) is helping scientists screen for potential medicines in new ways.
With larger, traditional molecules, pharmaceutical scientists use high-throughput screening (HTS)—such as some spectroscopic readouts from an enzyme-based assay—on libraries of a million or more components in search of new drugs. With smaller chemical fragments, screening only a few thousand of them often turns up a molecule that inhibits the target, although probably weakly. That inhibitor can then be optimized—made into a bigger molecule— to attack that target more strongly.
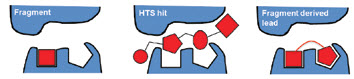 Fragments can fit target sites more precisely (left) than larger molecules found in high-throughput screening (middle), and then a fragment can be built into an even more tightly binding lead candidate (right).Image courtesy of Astex PharmaceuticalsThe compounds in a fragment-based screening library are 150–200 Daltons, compared to 400–500 Daltons for an HTS library. “With the smaller compounds,” says David Rees, chief scientific officer at Astex Pharmaceuticals (Cambridge, UK), “you can do a better job of screening all possible structures that might be available, because the total number of structures increases exponentially as molecular weight increases.” With the fragments, there are fewer possible structures to test.
Fragments can fit target sites more precisely (left) than larger molecules found in high-throughput screening (middle), and then a fragment can be built into an even more tightly binding lead candidate (right).Image courtesy of Astex PharmaceuticalsThe compounds in a fragment-based screening library are 150–200 Daltons, compared to 400–500 Daltons for an HTS library. “With the smaller compounds,” says David Rees, chief scientific officer at Astex Pharmaceuticals (Cambridge, UK), “you can do a better job of screening all possible structures that might be available, because the total number of structures increases exponentially as molecular weight increases.” With the fragments, there are fewer possible structures to test.
In a 2017 issue of the Journal of Medicinal Chemistry, David Bailey, director at IOTA Pharmaceuticals (Cambridge, UK), and his colleagues wrote: “The early-stage hits generated from fragment-based programs may have more favorable physicochemical properties compared [with] those resulting from high-throughput screening.” The authors added that more than 30 compounds from FBDD are in clinical trials, and two—vemurafenib and venetoclax—are on the market.
The current use of FBDD is just a start. It provides a wide range of benefits that will likely drive this approach into more pharmaceutical science ahead. In fact, scientists are only just exploring some of the ways to apply this approach.


OPTIMIZING OPPORTUNITIES
“It’s been suggested that the quality of interactions that fragments make with their target is potentially better than large, complex molecules,” says Seth Cohen, professor of chemistry and biochemistry at the University of California, San Diego. Cohen is also a co-founder of Forge Therapeutics, a start-up company that utilizes FBDD for drug discovery.
In part, smaller molecules bind in a more idealized manner. A larger molecule has to accommodate multiple interactions at once, which makes for suboptimal contacts. “Fragments can make key interactions with the active site, so you can see how it fits and then elaborate on that while preserving that original interaction,” Cohen explains. “You build your molecule piece by piece.” In addition, fragments can deliver higher hit rates, especially for more difficult drug targets, Rees notes.
At Astex, scientists used FBDD to discover an inhibitor of two proteins involved in cell death, and this spurred the development of ASTX660—an Astex molecule that is “currently being evaluated in a Phase 1/2 study in patients with advanced solid tumors and lymphomas,” Rees says.
Instead of searching fragment libraries with HTS, scientists screen them with biophysical methods, like mass spectrometry, nuclear magnetic resonance, surface plasmon resonance, or X-ray crystallography.
“These methods are used because fragment molecules generally bind rather weakly and are so small that it’s hard to predict where they might bind,” Cohen explains. So the analytical techniques must provide more resolution.
As reported in a 2017 issue of the Journal of Medicinal Chemistry, Cohen, along with Walter Fast of The University of Texas at Austin and their colleagues, applied FBDD to find new inhibitors of New Delhi metallo-β-lactamase-1 (NDM-1), which reduces the efficacy of β-lactam antibiotics. As Cohen explains, “We ran a screening campaign using libraries developed in our lab, and we found a novel core scaffold that bound to the active site of NDM- 1.” The team confirmed the binding and developed that into a lead-like molecule.
There are few areas of pharmaceuticals that are in as much need as antibiotic development. With increasing resistance among infectious diseases, any technique that turns up more lead molecules should be explored.

Advanced Lab Management Certificate
The Advanced Lab Management certificate is more than training—it’s a professional advantage.
Gain critical skills and IACET-approved CEUs that make a measurable difference.
ADDING IMPROVEMENTS
Although FBDD already provides pharmaceutical scientists with more capabilities, this technology can be further refined. For example, Astex focuses on only FBDD, but it collaborates often with pharmaceutical companies that use HTS. “That’s a particularly powerful method,” Rees says.
To make FBDD’s technology even more powerful, Rees sees several technologies that could be improved. For one thing, better technology to screen for fragments that bind targets even more weakly would help scientists find new potential lead candidates, and then their binding strength could be improved with further molecular modifications. “Plus, an increasing number of X-ray crystallography structures for potential drug targets would make it possible to see how small fragments bind the proteins,” he says. “Then you can grow the fragment into a lead in a very efficient way.”
As Cohen concludes, “A fragment-based approach is a good area to help people find new compounds that, maybe, they couldn’t find by other methods.”






