The Coherent X-ray Imaging (CXI) experimental station at SLAC's Linac Coherent Light Source X-ray laser, shown here, is specially designed for protein crystallography experiments. A new software tool, called Prime, is designed to reanalyze and improve LCLS crystallography results.Photo courtesy of the SLAC National Accelerator LaboratoryUnderstanding how our biology works at the atomic scale is a key to understanding and treating disease. But seeing the structure of proteins, the body’s microscopic machines, is a “big” problem: it requires big science facilities, generates big data – enough to fill tens of thousands of DVDs – and can require big research collaborations.
The Coherent X-ray Imaging (CXI) experimental station at SLAC's Linac Coherent Light Source X-ray laser, shown here, is specially designed for protein crystallography experiments. A new software tool, called Prime, is designed to reanalyze and improve LCLS crystallography results.Photo courtesy of the SLAC National Accelerator LaboratoryUnderstanding how our biology works at the atomic scale is a key to understanding and treating disease. But seeing the structure of proteins, the body’s microscopic machines, is a “big” problem: it requires big science facilities, generates big data – enough to fill tens of thousands of DVDs – and can require big research collaborations.
Now, a team led by Stanford scientists has created software that tackles the big data problem for X-ray laser experiments at the Department of Energy’s SLAC National Accelerator Laboratory. The program allows researchers to tease out more details while using far fewer samples and less data and time. It can also be used to breathe new life into old data by reanalyzing and improving results from past experiments at the Linac Coherent Light Source (LCLS) X-ray free-electron laser, a DOE Office of Science User Facility.
The tool, which will become publicly available, works by analyzing partial, X-ray-produced images of crystallized protein structures, known as diffraction patterns, that might otherwise be discarded and comparing them with known data to fill in the blanks and produce a more complete picture of these biomolecules. When applied to a whole set of data, this can reveal new structural details.
“We have reduced the required amount of diffraction data that’s needed to get a clearer picture of crystal structures and the time it takes to get a full structure of a biomolecule,” said Axel Brunger, professor and chair of Molecular and Cellular Physiology at Stanford and a member of the photon science faculty at SLAC, who helped to create the new software tool, called Prime.
“This is especially important because LCLS is in such high demand,” he added, as fewer than 1 in 4 experimental proposals at LCLS can be approved.
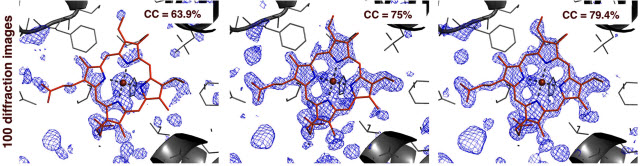 These three computerized renderings, based on an analysis of data from an experiment at LCLS, show how a software tool called Prime can aid in determining the 3-D structure of biomolecules. The image at right shows Prime-refined data for a sample of just 100 X-ray-produced images, called diffraction patterns, of a crystallized form of myoglobin, a protein found in muscle tissue. The image at left shows a simple merging and averaging of the same data, while the middle image shows partial image-correction using another method. The blue in the images represent a 3-D map of electron density in the sample, while the red shows the molecular structure. The "CC" value represents the "correlation coefficient," a measure of data quality in crystallography experiments, with a higher percentage resulting in a higher-quality structure.Images courtesy of Stanford UniversitySome biological experiments at SLAC’s LCLS have consumed millions of samples in the form of microscopic crystallized biomolecules, produced loads of data, and required a lot of computing power and data analysis. Because of this complexity, LCLS experiments often include dozens of collaborators from research centers around the globe, including scientists with data expertise.
These three computerized renderings, based on an analysis of data from an experiment at LCLS, show how a software tool called Prime can aid in determining the 3-D structure of biomolecules. The image at right shows Prime-refined data for a sample of just 100 X-ray-produced images, called diffraction patterns, of a crystallized form of myoglobin, a protein found in muscle tissue. The image at left shows a simple merging and averaging of the same data, while the middle image shows partial image-correction using another method. The blue in the images represent a 3-D map of electron density in the sample, while the red shows the molecular structure. The "CC" value represents the "correlation coefficient," a measure of data quality in crystallography experiments, with a higher percentage resulting in a higher-quality structure.Images courtesy of Stanford UniversitySome biological experiments at SLAC’s LCLS have consumed millions of samples in the form of microscopic crystallized biomolecules, produced loads of data, and required a lot of computing power and data analysis. Because of this complexity, LCLS experiments often include dozens of collaborators from research centers around the globe, including scientists with data expertise.
By applying Prime to earlier LCLS results, researchers produced a better 3-D map of the density of electrons in myoglobin, a protein present in muscle tissue. These maps allow researchers to determine the position of individual atoms in a protein. Also, they produced a higher-quality map of a bacterial enzyme using a randomly selected test batch of just 100 diffraction images from a full data set. The tool is described in the March 17 edition of the science journal eLife.
Prime, which stands for “post-refinement and merging,” could allow researchers to compress some experiments that used to take several days into hours or even minutes, greatly expanding the capacity for biological studies at LCLS while reducing the data deluge. It could make experiments more accessible to researchers who otherwise lack the special expertise to analyze and interpret LCLS results, and consume gigabytes rather than terabytes, or thousands of gigabytes, of data.
“Some LCLS experiments had required a tremendous amount of sample, and that was a huge limitation,” said William Weis, chair of the Department of Structural Biology at the Stanford School of Medicine and chair of the photon science faculty at SLAC, who also guided Prime’s development.
“It restricted a large number of experiments from even being attempted. With Prime, you don’t need as much redundant data,” Weis said, which should prove useful for studying membrane proteins that are popular targets for new drug development, for example, but can be challenging to produce in large quantities.
 Tiny crystallized biomolecules in a liquid solution (right) are streamed into X-ray laser pulses (shown as a white beam) in this illustration of crystallography at SLAC's Linac Coherent Light Source X-ray laser. Stanford University researchers have led the development of a new software tool that can reproduce 3-D molecular structures (left) using fewer X-ray-produced images of crystal samples.Image courtesy of the SLAC National Accelerator LaboratoryThe practice of reanalyzing old data with new techniques has gained momentum across many fields with the increasing supply of big data and computing power. Reanalysis has been particularly popular in the field of particle physics, where experiments can produce massive data sets and virtual “needles in the haystack,” in the form of rare particle events, can be the key to new discoveries.
Tiny crystallized biomolecules in a liquid solution (right) are streamed into X-ray laser pulses (shown as a white beam) in this illustration of crystallography at SLAC's Linac Coherent Light Source X-ray laser. Stanford University researchers have led the development of a new software tool that can reproduce 3-D molecular structures (left) using fewer X-ray-produced images of crystal samples.Image courtesy of the SLAC National Accelerator LaboratoryThe practice of reanalyzing old data with new techniques has gained momentum across many fields with the increasing supply of big data and computing power. Reanalysis has been particularly popular in the field of particle physics, where experiments can produce massive data sets and virtual “needles in the haystack,” in the form of rare particle events, can be the key to new discoveries.
Prime’s creators were inspired by a data-processing technique for diffraction data developed in the 1970s for X-ray sources called synchrotrons. It allowed researchers to map the structure of hard-to-study virus samples by compiling and analyzing a collection of incomplete diffraction data sets from individual crystals. Those partial data sets were compared to other data sets in order to obtain more complete data and refine the results.
“Even though the principal ideas were developed in the ’70s, this particular application required us to rewrite everything,” Brunger said, because of the unique properties of LCLS. In many biomolecular crystal experiments at LCLS, for example, the crystals are tumbling randomly when hit by X-rays, rather than individually and precisely rotated in the X-rays as they are at synchrotrons.
Brunger and Weis said several teams have already expressed interest in reanalyzing past diffraction data from LCLS experiments with Prime, which they said could lead to new structural insights.
In addition to Stanford and SLAC, researchers participating in the development of Prime were also from the Howard Hughes Medical Institute at Stanford, Lawrence Berkeley National Laboratory and Janelia Research Campus. The work was supported by the National Institute of General Medical Sciences, Howard Hughes Medical Institute and the U.S. Department of Energy.